| Revision as of 14:41, 8 September 2020 editDouble sharp (talk | contribs)Autopatrolled, Extended confirmed users, Page movers, File movers, Pending changes reviewers102,081 edits →The simple criterion for element placement: +1← Previous edit | Revision as of 14:46, 8 September 2020 edit undoDouble sharp (talk | contribs)Autopatrolled, Extended confirmed users, Page movers, File movers, Pending changes reviewers102,081 edits →Acknowledgements: having leafed through Scerri's book at a bookstore recently, I add a small bitNext edit → | ||
| Line 1,481: | Line 1,481: | ||
| Sc-Y-La is not taboo, it is a real secondary relationship, just like B-Al-Sc. That's all it should be, I claim. One should not try to promote it beyond what it really is, as that way lies inconsistency. | Sc-Y-La is not taboo, it is a real secondary relationship, just like B-Al-Sc. That's all it should be, I claim. One should not try to promote it beyond what it really is, as that way lies inconsistency. | ||
| ==Conclusion== | |||
| The physicists have been explaining the periodic system for ages: deriving Madelung's rule (Klechkovsky already did it). Since Hamilton's paper of 1965, they have been explicitly telling chemists about the mistake in group 3 (Lu should be there, definitely not La). Now there is between physicists and chemists regarding the verification of the superheavies. | |||
| It seems to me that physics has through its explanatory power earned its right to assert stronger custody of the periodic table. . | |||
| (P.S. IRL I am neither a physicist nor a chemist. Even if I do side with the physicists. XD) | |||
| As Scerri is now in favour of the left-step table with Sc-Y-Lu and He-Be (see ''The Periodic Table: Its Story and Its Significance'', 2nd ed., p. 403), and he is the chair of on group 3, I am cautiously hopeful that the story of what I consider the erroneous placement of three elements on the table (helium, scandium, and yttrium) will, soon enough, God willing, have a happy ending. | |||
| ==Acknowledgements== | ==Acknowledgements== | ||
Revision as of 14:46, 8 September 2020
Caveat lector! This is not so much an organised document as it is a periodicity commonplace book. The author also thinks a rewrite is needed but is on a Wikibreak. So it is left up for now in the current state.
TL;DR summary: Lu under Y, He over Be.
The simple criterion for element placement
I have just one simple criterion for element placement (you can't get much simpler than that):
An element is placed in the periodic table according to its number of valence electrons and the valence orbitals they can go into.
To put it explicitly: if you have 1 or 2 valence electrons, you go in the obvious column of the s block, as they must be s electrons and those are the ones filled up front that will survive the whole row or nearly so (other subshells may be active). We explained why above. And if you have more (n where n > 2), you go into the block corresponding to your valence orbital of highest angular momentum, and in the (n − 2)nd column of that block.
This is essentially an even more condensed version of Jensen's criteria (cite him):
- Assignment of the element to a major block based on the kinds of available valence electrons and/or valence vacancies (i.e., s, p, d, f, etc.).
- Assignment of the element within a given block to a particular group based on the total number of available valence electrons.
- Verification of the validity of the resulting block and group assignments through the establishment of consistent patterns in overall block, group and period property trends.
- Verification that the elements are arranged in order of increasing atomic number as required by the periodic law.
We will now see that this gives relevant results.
(Just looking at chemistry alone won't cut it, because then we'll always be stuck in indecision over whether Be and Mg belong with group IIA or IIB, or whether B and Al belong with group IIIA or IIIB. To say nothing of the La vs. Lu issue for group 3, which is hopeless to resolve without considering electronic structure. Electronic structure does control chemistry, but the relationship is not simple at all.)
Of course this is kind of problematic in late 7th period and early 8th period because of weirdly timed drowning of s and p1/2 subshells. (Radon is also maybe a problem, but maybe +8 does exist after all. Francium and radium may have some 6p cohesive strengthening of bond.) However, I guess the tail need not wag the dog here. Instead of failing at random times for no good reason, like ground state gas phase electron configurations, this has a very obvious reason behind its failure. The effects of special relativity have started to be significant. So, what we do instead is say "okay, everything is fine for the non-relativistic part of the table, which may be pushed to at least most of the first six rows; after that we do the best we can".
Why such a criterion?
I quote Eric Scerri, The Periodic Table: Its Story and its Significance (2nd edition, p. 127):
| “ | For Mendeleev, the element was an entity, which was essentially unobservable but formed the inner essence of simple bodies. Whereas a particular “element” was to be regarded as unchanging, its corresponding simple body aspect could take many forms, such as charcoal, diamond, and graphite, in the case of carbon. In this respect, Mendeleev may be thought of as upholding the ancient philosophical tradition regarding the nature of elements as bearers of properties. Mendeleev’s genius now lay in recognizing that just as it was the “element” that survived intact in the course of compound formation, so atomic weight was the only quantity that survived in terms of measurable attributes. He therefore took the step of associating these two features together. An element (basic substance) was to be characterized by its atomic weight. In a sense, an abstract element had acquired a single measurable attribute that would remain unchanged in all its chemical combinations. Here is a profound justification for using atomic weight as the basis for the classification of the elements, quite unlike anything produced by other discoverers or precursors of the periodic system. How else is one to make sense of Mendeleev’s otherwise rather naive-sounding claim that he had realized the need to order the elements according to atomic weight, given that others had done so before him? The point is that he was providing a detailed account of why this was the correct approach to take. | ” |
| — Eric Scerri | ||
The valence orbitals, being a consequence of the atomic number, strike me as a reasonable route to take here. They are the foundation of chemistry. Nothing else is fundamental enough (now that we know atomic weights won't cut it). And this must be applied for all elements, as Mendeleev believed that the periodic table should not admit any exceptions.
Chemistry and physics alone will be suggestive but cannot cut it, looking at the immense physical variation in F-Cl-Br-I-At-Ts, or the immense chemical variation in C-Si-Ge-Sn-Pb-Fl. Only valence orbitals will allow us to explain all of those under one roof.
So, just what is a valence orbital?
I suggest the direct idea: look for bonding AOs in a survey of compounds. That's the ideal even though very intense. Then I'm quite sure those things will vanish and you'll get what is required. Which is good because that is the most natural meaning of "valence" in the first place. In some cases you must be careful such as dual localised-delocalised 4f. ;)
From what I understand especially buried subshells tend to come out more when bonded to electronegative substituents, that seems to be accurate for 3d (ZnF2 has significant 3d involvement, ZnS not really), and 4f (YbO has it for 4f, YbS can be neglected and be okayish). Those are the deadline emements, Ga 3d is core, Lu 4f is core. Somehow 7s is restored by fluoride ligands even in NhF4, the effect seems real even if I do not understand why yet. Same thing with lead if I read this paper correctly.
Do not be fooled by oxidation states! Zn 3d is active despite no oxidation states past +2 being known yet. Organometallic compounds of Pb, Bi, likely Po in their group oxidation states are quite stable but 6s is almost totally nonbonding!
So maybe for definiteness and ease we say, good rule of thumb if you have no time to check everything is to check orbitals in use in fluorides and oxides of maximum oxidation state. If it's not used there, it will probably not be used anywhere. I guess I do not have to say that we must only use chemically real oxidation states. No CsF3 please. ;) Of course for contentious cases like La and Ac you must do more to really prove it. ;)
I trust DA will explain to me if this does not work. ;)
Now, this is still a bit complicated, so it is good that there is a proxy. You know those configurations of single atoms? Well, just look for the excited states of the atom and (for metals) some cations, up to 10 eV (~ limit of chemical bond energy), those will give relevant configurations. As a bonus the subshells like La 4f, Ac and Th 5f will fall down even further and become obviously valent because of less screening from nucleus. Now that's something that can be done very easily.
BTW, you don't really need sanity for noble gases, just use charged compounds. HeH is well-known. So there are even fewer exceptions than I thought, just look at other compounds.
The table of idealised electron configurations
Given how the "outer shell" of s+p is important and generally acts together, we may dare to suggest that although the n+l rule seems to work well (it has even been derived by Klechkovsky and D. Pan Wong), the important split is actually from the noble gas to the alkali metal, so that we should have an LSPT with the last two columns moved to the front. There is a big gap from 4p to 5s, but 4s and 4p act together despite different n+l values, so I suggest:
1s; 2s 2p; 3s 3p; 4s 3d 4p; 5s 4d 5p; 6s 4f 5d 6p; 7s 5f 6d 7p; 8s etc.
with the semicolons marking the more chemically relevant breakpoints. As each noble gas core gets less noble than the previous one, this approximation loses some relevance, but it is still largely right (you can see some cracks with 5p involvement in the sixth row, but they're still not the main thing: doi:10.1039/c3cp50717c).
This is quantitatively weakened at the end of period 7 and going into period 8 due to relativistic effects (s is lowered but p3/2 is raised, so the gaps quickly stop being big in the right way), but amazingly the qualitative idea is still all right: it's just that near the end of period 7, the important s-orbital becomes 8s instead of 7s, and in period 8 it changes from 8s+8p1/2 to 9s+9p1/2, but that results in non-relativistic gap types. These happen over time, so what we get is interesting. Valency and maximum oxidation state of flerovium, moscovium, and livermorium are two too low (you see this starting already as it is the favoured oxidation state already for in higher elements: antimony prefers +3 to +5, bismuth has strong preference, moscovium likely has no 5) because 7s is inactive whereas 7p1/2 is active, while for tennessine and oganesson they are four too low because 7p1/2 has also been drowned. Same thing happens in 5g elements: we choose to extend the g block four elements, (otherwise it is difficult to make the fit work, and the g electrons seem active that far). So instead of early 6f elements having "two too low" oxidation states due to 8s drowning, they end up as two too high (probably E143 can go up to +5 state with valence configuration of (6f 7d 8p1/2) which is just weird under lanthanum and actinium), but at the end they end up just right with E155 and E156 as good homologues of mendelevium and nobelium respectively because 8p1/2 has also drowned by then. However, because this is kind of plain weird, one can also suggest Droog Andrey's original template (please move helium over to the left) to get the point: E139-E156 are kind of like 18-element actinide series, E121-138 are kind of like 18-element lanthanide series, they correlate because of filled 5g subshell added, but due to the drowning periodicity is not good. I do not know which is better, the problem with drowning is too bad. ;)
{kind=link}
Maybe homology of E157-E172 to Y-Xe is just an accident, or a manifestation of periodicity among later elements: so far, we don't know. And I guess we will not have an idea until calculations are done for period 9 elements, assuming those will live long enough to have real chemistry.
However, please just also note that the s orbitals sticking around to the end of the row was itself the exception from n+l, so if they don't manage to get to the end in row seven it simply means the exception has an exception, no big deal already. Same thing with 7p1/2, it's another of those relavistically contracted ones. So I argue that you can interpret this as working all the way: period 8 does the same thing, so much so that the ninth of these subshells flies in and replaces the eighth, so periodicity is delayed four elements from Aufbau. All is clear anyway, it is not really an exception. In fact by this logic so is the exception where some 6p involvement appears in francium and radium not really an exception as it is already an exception from an old exception. Maybe to eliminate exceptions we can argue for the original Janet table but I must say I think the original exception is exceptional enough and the exceptions from it only rare exceptions that it makes sense to make only the first exception. And now I have saturated you semantically with the word "exception". ;)
Groups 1 and 2 deserve comment due to early pre-d involvement, because the next higher subshell must have angular momentum above 0. This is an s-block problem, as l "resets" after the s-subshell, but periodicity resets before it: indeed, this is behind why the s-block is the only one to have no double periodicity. We note the following:
- There is already p-involvement due to hybridisation in Be and Mg, for instance, so it's not terribly different from d-involvement in Ca through 120. (Maybe we can expect pre-f character in Ba? Best of all, E173 has predicted "pre-g" involvement with its 6g configuration.) The s-block fraternises with the next set of subshells with the next value of n+l, rather than going with the one it theoretically should show in. That's why I still insist on starting with alkali metals and ending at noble gases, rather than the LSPT arrangement.
- For confirmation we go down to the next step and count the number of available valence electrons: He, Be and Mg have two, as do Ca through 120, but Zn through 166 have twelve.
Most amusingly, this suggests that H and He are the only pure s-elements... Anyway, ignoring the s-block for now, you can see from the valence subshells the source of the p-block octet rule, the d-block 18-electron rule, and the f-block 32-electron rule. (Maybe there will be a 50-electron rule somewhere. More likely for 6g than 5g, I expect.) The s-block again is poised ambiguously; the pure s-elements H and He use a duet rule, but after that we have octets and some inklings of octodecets for Ca, Sr, and Ba. These rules are why most of this is uncited as common knowledge as anyone can think of lots of examples of octets and octodecets respectively for the p and d blocks. For some other weird ones:
- Lithium, 2p as valence orbitals (back-donation in MeLi) https://pubs.acs.org/doi/full/10.1021/om950966x?src=recsys
- Beryllium, 2p: common knowledge (sp hybridisation, BeCl2) + sp in condensed phase
- Sodium, 3p: I admit, I am still looking
- Magnesium, 3p: also common knowledge, + (metal ions)
- Heavy group 1 and 2 (K-Cs, Ca-Ba), d-involvement: http://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.600.2560&rep=rep1&type=pdf (it increases down the table; for K it is minor, but at Cs it is approaching that of Ca)
The precise composition of "all higher" may change as the table is descended, bearing in mind the increasing s-p gap down the table and the decreasing s-d gap. The important thing is that the s-block is an exception to the principle that the characteristic subshell has the highest angular momentum among all valence ones. That is only true for hydrogen and helium.
- 4f band for Cs and Ba under pressure: here
This is an annoying symmetry break: normally you would expect the new orbitals to come in, but for Cs and Ba they don't at normal pressure. The 4f collapse is fast. At barium it is still hydrogen-like and far from the nucleus; by lanthanum it suddenly collapses to just above 5d (not hydrogen-like anymore); by cerium they are about equal; by praseodymium and neodymium it is below; and then you get to approximately the state in the later lanthanides where 4f is a "reserve" orbital where electrons are promoted out of to participate directly in the bond. The real symmetry break in the table is the s block: everyone else ends involvement of the characteristic orbital on time...
We also have to note that very deeply buried valence subshells suffering primogenic repulsion (4f, 5g) have a special rôle as "reserve valence" subshells. In the early part of a series they may be more effective paradoxically when their collapse is still waiting (cite that paper about cubic complexes in La and Ce rotating to squre antiprismatic later in the 4f row). Although they do not participate so directly in the bonding normally, they are a place valence electrons can move into and out of, coming out to participate chemically and causing an interplay of 4fn and 4f(n+1) configurations. (Cite Ionova) This situation exists from La to Yb, and not at Lu. The "delayed collapse" effect we see in ground-state configurations for La-Ce, Ac-Th-Pa, E121-E126 means absolutely nothing: it is common for all heavy elements (see the collapses for 6d and 7d), and should not be taken as something special for the f and g blocks when chemically delayed-collapse elements are actually more likely to use those orbitals than the ones that appear later in the first row (compare what happens before and after Ce when 4f collapses; likely we are in the same situation before and after E125 when 5g collapses).
The point is that valence orbitals do something for the bonding. You cannot reduce it to just the "characteristic subshell" that names the block, because everyone knows that the others are also doing something (whence the octet and 18-electron rules for example), and hybridisation is something utterly normal throughout the entire periodic table.
On d-participation in Zn, Cd, Hg as cohesively strengthening the bonding, I quote Droog Andrey: "The equilibrum internuclear distance in ZnCl2 molecule is 2.07Å, suggesting that chlorine atom penetrates quite deep into the 3d subshell of zinc; the corresponding value for LuCl3 is 2.4Å, but 4f subshell in lutetium is smaller than 3d subshell in zinc." (Archive 33 of WT:ELEM)
On d-participation in f-elements: this is kind of common knowledge for the lanthanides, for which 5d and 6s contribute far more direct bonding than 4f usually. Also, for lanthanides and actinides an important factor is promoting an electron out of the f shell into the d one where it becomes directly valent. p usually has minor contribution, but it can sometimes be surprisingly high (e.g. Ac2 dimer).
Once that is settled, homology works perfectly up to period 7. In period 8 it is straining but still holding the fort. I insist to not treat the predicted elements differently. Chemically, we know exactly as much about E125 as we do about Mt: nothing in practice, some stuff in predictions. So we may begin the drawing of period 8 early according to the state of the art, since ever since element 102 (No) was discovered, physics came before chemistry. Otherwise the periodic table ends at hassium today. ^_^
I have dared to draw in a period 9 just as a soft prediction, just following the periodic law. Time will tell if this is prescient or hopelessly naïve. Since Glenn T. Seaborg drew a table up to E168 and expected Aufbau to hold, I am fearless! ^_^
| (s++) | (s++) | (dsp) | (dsp) | (dsp) | (dsp) | (dsp) | (dsp) | (dsp) | (dsp) | (dsp) | (dsp) | (sp) | (sp) | (sp) | (sp) | (sp) | (sp) | |
| Is | IIs | IIId | IVd | Vd | VId | VIId | VIIId | IXd | Xd | XId | XIId | IIIp | IVp | Vp | VIp | VIIp | VIIIp | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H | He | 1s | ||||||||||||||||
| Li | Be | B | C | N | O | F | Ne | 2s2p | ||||||||||
| Na | Mg | Al | Si | P | S | Cl | Ar | 3s3p | ||||||||||
| K | Ca | Sc | Ti | V | Cr | Mn | Fe | Co | Ni | Cu | Zn | Ga | Ge | As | Se | Br | Kr | 3d4s4p |
| Rb | Sr | Y | Zr | Nb | Mo | Tc | Ru | Rh | Pd | Ag | Cd | In | Sn | Sb | Te | I | Xe | 4d5s5p |
| Cs | Ba | Lu | Hf | Ta | W | Re | Os | Ir | Pt | Au | Hg | Tl | Pb | Bi | Po | At | Rn | 5d6s6p |
| Fr | Ra | Lr | Rf | Db | Sg | Bh | Hs | Mt | Ds | Rg | Cn | Nh | Fl | Mc | Lv | Ts | Og | 6d(7→8)s7p (*) |
| 119 | 120 | 157 | 158 | 159 | 160 | 161 | 162 | 163 | 164 | 165 | 166 | 167 | 168 | 169 | 170 | 171 | 172 | 7d(8→9)s(7→8→8/9)p (*) |
| 173 | 174 | 211 | 212 | 213 | 214 | 215 | 216 | 217 | 218 | 219 | 220 | 221 | 222 | 223 | 224 | 225 | 226 | 8d(10→11)s(10→9/11)p ? |
| (fdsp) | (fdsp) | (fdsp) | (fdsp) | (fdsp) | (fdsp) | (fdsp) | (fdsp) | (fdsp) | (fdsp) | (fdsp) | (fdsp) | (fdsp) | (fdsp) | |||||
| IIIf | IVf | Vf | VIf | VIIf | VIIIf | IXf | Xf | XIf | XIIf | XIIIf | XIVf | XVf | XVIf | |||||
| La | Ce | Pr | Nd | Pm | Sm | Eu | Gd | Tb | Dy | Ho | Er | Tm | Yb | 4f5d6s6p | ||||
| Ac | Th | Pa | U | Np | Pu | Am | Cm | Bk | Cf | Es | Fm | Md | No | 5f6d7s7p | ||||
| 143 | 144 | 145 | 146 | 147 | 148 | 149 | 150 | 151 | 152 | 153 | 154 | 155 | 156 | 6f7d9s8p1/2 | ||||
| 197 | 198 | 199 | 200 | 201 | 202 | 203 | 204 | 205 | 206 | 207 | 208 | 209 | 210 | 7f8d11s10p1/2 ? |
| (gfdsp) | (gfdsp) | (gfdsp) | (gfdsp) | (gfdsp) | (gfdsp) | (gfdsp) | (gfdsp) | (gfdsp) | (gfdsp) | (gfdsp) | (gfdsp) | (gfdsp) | (gfdsp) | (gfdsp) | (gfdsp) | (gfdsp) | (gfdsp) | (gfdsp) | (gfdsp) | (gfdsp) | (gfdsp) | |
| IIIg | IVg | Vg | VIg | VIIg | VIIIg | IXg | Xg | XIg | XIIg | XIIIg | XIVg | XVg | XVIg | XVIIg | XVIIIg | XIXg | XXg | XXIg | XXIIg | XXIIIg | XXIVg | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 121 | 122 | 123 | 124 | 125 | 126 | 127 | 128 | 129 | 130 | 131 | 132 | 133 | 134 | 135 | 136 | 137 | 138 | 139 | 140 | 141 | 142 | 5g6f7d8s8p1/2 |
| 175 | 176 | 177 | 178 | 179 | 180 | 181 | 182 | 183 | 184 | 185 | 186 | 187 | 188 | 189 | 190 | 191 | 192 | 193 | 194 | 195 | 196 | 6g7f8d10s10p1/2 ? |
s++ means "including at least some of the higher-l orbitals in the same row". I don't posit any for H and He (pure s-elements). For Li and Be, it means 2s+2p; for Na and Mg, 3s+3p; for K and Ca; 4s+3d+4p; etc. Unfortunately my old conjecture for 4f and 5f to join in the fun has not proved a happy one except under pressure (where I won't go because then yttrium maybe has 4f involvement(!)), but then again the s-block has already been the symmetry break.
(*) Due to relativistic effects, one may consider 7s7p to become (7→8)s7p for the last four elements of the period, and 8s8p to really be 8s(7→8)p in the beginning for E119. Also, flerovium is problematic as 8s is probably not low enough but 7s is probably not high enough. (Nh may have 7s restored in anionic complexes where +3 is stabilised, but Fl +4 is probably too oxidising even for fluorine. I wonder if we could force it to happen with teflates...) This is a slight perturbation because of the lowered 7p3/2-8s gap thanks to relativistic effects; at this point nonrelativistic subshells are no longer a good approximation and the Madelung rule is starting to need the 8th-period amendments where the s and p subshells start splitting and changing in energy. The bigger diagonal pattern is still relatively all right when it comes to predicting the order of blocks, though: there still never appears any such thing as a "split" block like the Sc-Y-La form would have us believe in, all the way to E172 (and probably E184 from what glimpses we have so far had of period 9).
Notice that the g-block appears to be 22 rather than 18 columns wide. That's all right. The important thing is that these are the elements where 5g is a valence subshell. The boundary between 5g and 6f is a little fuzzy, and there are some grounds for considering as a "secondary periodicity" E139-E156 as homologues of E121-E138 with a g-shell added, but the lack of radial nodes in 5g and their presence in 6f makes a significant difference (5g is a bit like actinides up to Pu, then they stay around there creating very similar elements like lanthanides, but then 6f increases oxidation states even further maybe quite past +8 before dropping down a bit like actinides). But we should stress that this 8th period is only preliminary (and this 9th period is less than that). Predictions have varied over time for period 7 and they should do it for period 8 as well. What we have here is a preliminary "scaffolding" idea, since we will surely have to put the elements up before we can chemically investigate them. When we do so, something may change. The problem is that 6f is fuzzy because 8p1/2 gives way to 9p1/2 in the middle of it, leading to the oxidation states problem. But, well, not much else is better.
I dislike Pyykkö's extended table, in particular. Whatever happened to the periodic law demanding that elements appear in atomic number order? Whatever happened to the role of the number of valence electrons, which is trampled on when you put E155-E164 as your d block in stead of E157-E166? And what exactly do E139 and E140 have to do with the boron and carbon groups? And why split E167-E168 to E169-E172 and create the false impression that there is some difference between the nearly degenerate 9p1/2 and 8p3/2 sub-subshells? Fricke is marginally better but still has the wrong d block. I strongly prefer this one (following Nefedov et al., and Kulsha). At least until we have complete calculations past E123, I guess. ^_^
In particular, note that in Lu 4f is a core subshell, no different from Hf. The drowning gets deeper and deeper as we continue down the 6th period, so there is a quantitative difference; but there is no qualitative difference.
Group names are shortened to refer to the characteristic subshell only, therefore anchoring ourselves straight to the main criterion. In fact, this is very similar to Wulfsberg's approach at the beginning of Principles of Descriptive Inorganic Chemistry (which indeed names the f-block columns as 3f through 16f; I use Roman numerals to make them not look like subshells), except that I consider the p electrons to be relevant in the other blocks too, and consider all orbitals possibly relevant for the s block. The fact that past VIII (and under extreme conditions, IX once) you don't ever see the group oxidation state being attained is not a problem: how big an oxidation state can get before the trend starts going down varies throughout the table (just compare the beginning of each period, you'll see +1 in period 1, +5 in period 2, +7 in periods 3 and 4, +8 in period 5 as maxima; not to mention the change between 4f and 5f), that's already one level deeper. They do turn out very similar to the Marks Brothers' group labels, but I think we can see that my approach is very different from theirs (I had seen their article, but I wasn't really thinking about it).
Another precedent is Shemyakin and Mashentsev
(I apologise for the lack of asterisks. My sole excuse is that once you have two footnotes, as happens once the g block opens, it gets really confusing. The 32-column version below, which has asterisks, should make it clear what I mean by this: the gluing must be done in the only way that preserves the increasing sequence of atomic numbers.)
Logical flow
To recap: the logical flow of discovering the criterion is that we start by observing what electrons are being used for chemistry, creating a list like so:
1s hydrogen (1 electron) 1s helium (2 electrons) 2s+2p lithium (1) 2s+2p beryllium (2) 2s+2p boron (3) 2s+2p carbon (4) 2s+2p nitrogen (5) 2s+2p oxygen (6) 2s+2p fluorine (7) 2s+2p neon (8) 3s+3p sodium (1) ... 3s+3p argon (8) ~3d+4s+4p potassium (1) ... 3d+4s+4p zinc (12) 4s+4p gallium (3) ... 4s+4p krypton (8) ~4d+5s+5p rubidium (1) ... 4d+5s+5p cadmium (12) 5s+5p indium (3) ... 5s+5p xenon (8) 5d+6s+6p caesium (1) 5d+6s+6p barium (2) 4f+5d+6s+6p lanthanum (3) 4f+5d+6s+6p cerium (4) ... 4f+5d+6s+6p ytterbium (16) 5d+6s+6p lutetium (3) 5d+6s+6p hafnium (4) ... 5d+6s+6p mercury (12) 6s+6p thallium (3) ... 6s+6p radon (8) 6p+6d+7s+7p francium (1?! oops) 6p+6d+7s+7p radium (2?! oops) 5f+6d+7s+7p actinium (3) 5f+6d+7s+7p thorium (4) 5f+6d+7s+7p protactinium (5) 5f+6d+7s+7p uranium (6) ... 5f+6d+7s+7p nobelium (16) 6d+7s+7p lawrencium (3) 6d+7s+7p rutherfordium (4) ... 6d+7s+7p copernicium (12) 7s+7p nihonium (3)
and simply arrange the pattern in a table. Note, the s block participation of higher orbitals is a bit inconsistent sometimes (clearest for p, and for d in Ca-Sr-Ba and Cs; for K and Rb it is weaker, and for f it is quite weak until pressure is applied), but then it is the one consistent exception. And that's how we find the pattern staring in our faces with just a few kinks near the end due to relativity.
The table, drawn according to those principles for the first 8 rows
Yes, I dare to claim that there is such a thing as the ideal table when it comes just to element placement based on chemistry and physics.
I decided to forgo categorisation by metallishness and use blocks only. That is the only thing that makes sense, please do not confuse elements with simple substances. ;)
| Periodic table (Large cells) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| s-element | g-element | f-element | d-element | p-element |
I would rather show the real 54-column table rather than specifying gluing rules, but then this would trail well off the page and be generally annoying to print. In the 18-column form: oh boy, you'd need a footnote under a footnote.
Now the acid test: has our criterion produced something that makes sensible trends appear? We see we did get them for group 3 (just examine Jensen's figure and paper), but we also seem to have a weird period 8 and a weird helium. Are those sensible or falsifiers? Remember, a scientific theory must be refutable in principle. Or else it explains everything and therefore can't be used to predict anything, as it gives no strong reason why A should happen instead of B. Karl Popper understood that very well.
This page used to have a big chunk of text explaining why Lu under Y is, IMHO, totally correct. It is now at User:Double sharp/Lutetium manifesto.
Helium
- I'm not happy with the exposition of this section now, but I also don't have time to fix it, so it will stand for now Double sharp (talk) 13:34, 13 August 2020 (UTC)
This started all really because of the Eka-Yttrium Holy War, but I realised my approach demands He-Be if you use it consistently. So there are only two consistent approaches that do not entail a double standard: either reject my approach, or embrace He-Be. I decided on the latter. Now I will explain why it is not chemical nonsense even though it kind of looks like it is at first glance. It was a surprise to be sure, but a welcome one. XD And it's really not just about helium, but about hydrogen too.
Often, at least hydrogen is left to float aimless over the table; sometimes, helium joins it (e.g. Greenwood and Earnshaw). This is basically tantamount to throwing up one's hands, excluding it from the periodic law (which demands periodicity in chemical properties for all elements), and explaining hydrogen in one of the following ways:
- Magic
- Eirin's shady new drug
- Yukari fooling around again
- Moriya Shrine Conspiracy
Templates like that are forbidden! The periodic law must be binding for every element, or it is not a law at all.
Just because hydrogen is in a singular position does not mean we cannot rationalise it as springing from something deeper that is after all not so different from the rest of the elements. We can analyse hydrogen and helium just the same using valence orbitals: the only such is 1s. What makes them so different from the other main group elements is that there is no higher subshell: normally s+p form a complete octet, as happens from Li-Ne and Na-Ar. But this is a reasonable case extrapolating backward from Ca-Ra which use d-orbitals (as sd hybridisation is getting better) and Be-Mg which use p-orbitals (with a lower sp hybridisation defect than anywhere else); here H and He are really pure s-elements. We are using electronic configurations to place them: H is s, closer to Li than anything else, and He is s, closer to Be than anything else. They have the wrong number of valence electrons to go over F and Ne, which would be something unprecedented in the table. So by valence orbitals it is a shoo-in: H over Li, He over Be.
As an aside I remark that due to H having one valence electron and one valence vacancy, the only elements it makes any sense put over are lithium (one valence electron, seven vacancies) and fluorine (seven valence electrons, one vacancy). Similarly He only makes sense over Be or Ne. I believe in "one element, one place", so I do not support it duplicated over both (unless in parentheses to show a secondary relationship, or in a Bayley-Zmaczyński-type arrangement). I did not think I had to mention this, but since there is already a proposal to put hydrogen over carbon...
So we have a triple status regarding why hydrogen and helium should look really weird:
- 1s is severely contracted and small to begin with;
- 1s has no shielding from the nucleus;
- there is no 1p subshell, so just like (K-Rb)-Cs and Ca-Sr-Ba are one or two electrons out of 18, but Li-Na and Be-Mg are one or two out of 8, so H is one out of two and He is two out of two. So the significance is different.
So of course there are no good heavier counterparts in the way that Na-Ar act like reasonable big sisters of Li-Ne. There can't be any, 1s stands alone if you look at the filling arrangement.
I now claim that this is not chemical nonsense. Well, I guess the regularities coming from H over Li are well known. So here are what He over Be gives you.
- Placement of elements is about electronic structure that explains chemistry, not about correlations in the final chemical and physical behaviour that is a lot of steps removed from the origins even if still related. Otherwise we have trouble explaining nitrogen and bismuth in the same group: typically metallic chemical properties are much much much more developed in bismuth than in nitrogen, and the oxidation states favoured are different (N: +5/-3, Bi: +3).
- Atomic properties show the trends (graphed in Grochala's article and here) where He-Ne fits quite badly but He-Be fits sensibly as a first-row anomaly (as well, if you graph the noble gas trend, Ne-Ar looks like the stereotypical first-row anomaly, and not He-Ne).
- The knock-on effects on the following elements. The He-Ne difference actually explains why Li, despite being not exactly the most reactive alkali metal (translation: it's the least reactive one), is actually the most electropositive one, because 1s shields so well from the nucleus to a level that 2p-6p cannot manage (10.1002/jcc.20522). He over Be emphasises the range of applicability of the "duet rule" for the filled 1s subshell, and by its placement it shows clearly that this is something different from the octet core of the other noble gases.
- The first four elements (H through Be) are all s elements; if we are serious about blocks as a guiding principle, we have to consider He-Be primary periodicity and He-Ne secondary periodicity, like H-Li vs. H-F, because H-Li and He-Be are pairs from the same blocks and H-F and He-Ne are not. Yes, it's true, we don't teach the blocks from the start, but we still use them as our guide to how to place elements despite that and I think making an exception for helium is not right. It smacks of special pleading and immediately opens questions for B-Al-Sc-Y-La, Ti-Zr-Ce-Th, Be-Mg-Zn-Cd-Hg, Ca-Sr-Yb, and other wonderful denizens of Pandora's box. I would rather keep the box shut with the lid of consistency, even if chemically strange-looking He over Be is the price for that. We can explain its really weird behaviour for an s element by saying that it has been impacted by (1) lacking any shielding from the nucleus and (2) lacking a p-subshell. This is exactly like hydrogen, which we place standardly in group 1.
- Helium stands in relation to beryllium more or less as hydrogen stands in relation to lithium, and the latter is more or less standard by now. So if you think the latter is all right, with a nonmetal heading group 1 with some incipient metallic properties, than the former doesn't look quite so strange anymore. Though I freely admit that incipient metallic properties of helium are even weaker than those of hydrogen, but at the very least the huge first-row anomaly it represents now looks like something regular rather than saying "hydrogen is our weirdo". The general s block trend is for chemical activity to increase down each group as the valence electrons get farther from the nucleus; well, H-Li and He-Be certainly fit that.
- I dislike having to cut off hydrogen (and maybe helium) from the periodic law. If it is supposed to be absolute, and we make an exception right at the very beginning, it smacks of special pleading again. OK, maybe we have some exceptional cases at the beginning, but they should form some part of the normal trend and just represent a coincidence of various different groups. You can think of it as something like an exceptional isomorphism going on here, to use a mathematical analogy.
- This fits neatly with the principle of first-element distinctiveness (that, according to Henry Bent, a long-term He-Be advocate, goes back to Mendeleev himself). And it also fits with Jensen's principle of first-row anomalies, s >> p > d > f. I find most of Bent's arguments (mostly involving stuff like atomic number triads etc.) unconvincing but this one is an exception because it corresponds to real chemistry. Whereas there is nothing really distinctive about helium over neon, not to mention how it breaks reactivity and electronegativity trends.
- With helium in group 2, every main group starts with nonmetal(s) and then gets metallised. The faster metallisation for the s block fits with Jensen's principle of first-row anomalies, too. And it makes sense: just like 2p, 1s is really really small, nuclear attraction is huge, so Kaupp's explanation for the electronegativity dropping precipitously down from 2p to 3p also applies just as well from 1s to 2s! It even applies better because 1s shields even better than 2p! Such a trend is seen for H-Li and He-Be, but H-F and He-Ne instead increase electronegativity going down! It also means that a small 1s or 2p element should be bad at multicentre bonding (and metallic bonding is kind of the generalisation as the number of centres goes to infinity).
- The regularity is then that as we progress down an s-block group, our valence set expands. In 1s it's out of 2 electrons; in 2s and 3s it's out of 8; in 4s and 5s it's out of 18. (Unfortunately expansion to 32 for 6s and 7s seems to need pressure.) So in some sense we should expect He-Be as something not too different in nature from Mg-Ca (where Ca starts showing some serious d involvement), even if it is obviously of a once-in-the-entire-table scale (because when else do you have a first-row anomaly in an odd period, therefore implying that there's no homologous period after it without the first-row anomaly)? The general situation (due to kainosymmetry – yes, that's also from Shchukarev) is for the even periods to behave differently from the odd ones, with period 1 standing apart from both. (Because even periods have an orbital without radial nodes that creates incomplete shielding. Even periods have stronger oxidisers and suffer more significant contraction effects, because their size increase from the previous period has been cancelled.) Well: period 1 is absolutely unique, as it's an odd period without a partner and with kainosymmetry. So, the bad resemblances of H-Li and He-Be are in fact totally regular. H-F and He-Ne, which look better at first glance, would actually be unexpected in their relative goodness. Although I'm aware that this argument is dangerously close to saying "He-Be is correct because it is wrong". XD
- As Henry Bent noted: 4f elements are very similar to each other, 3d are similar (but less so) especially late first row from Mn onwards where +2 starts dominating, then 2p are quite heterogeneous and 1s even more so (impressively for just two elements).
- The difference also explains why He is more reactive than Ne: Ne chemistry is thwarted by lone-pair repulsion (that affects F2 for instance, and is a reason why the 2p elements favour multiple bonding), and just like what happens with H (immense polarising power of a naked proton) and H (a rather diffuse anion because the lone proton cannot easily manage this much negative charge), any disturbance of the filled 1s subshell of helium instantly increases reactivity significantly, as Grochala noted in his PCCP paper on (HeO)(LiF)2 (supplementary information).
- The stability of He compounds in predicted Ng-Be bond-containing compound series (Ng = He, Ne, Ar, Kr, Xe) drops off the clear trend from Ne-Xe. Instead He appears near Ne, and sometimes the He compound is even less unstable than the Be compound. (Grandinetti p. 74) By changing the ligand you can change the order of He vs. Ne (nothing is said about Ne-Xe, presumably it follows the standard order throughout), which suggests that He is to some extent an accidental part of the noble gas trend. (Grandinetti pp. 103ff.)
- Theoretically predicted He bonds not only lack Ne analogues, but are also significantly analogous to Be.
I realise the last three are weak arguments because He compounds are quite outside the bounds of "normal chemistry", but the others are not.
I won't deny that at least partly my support of He-Be started as a provocation. Everyone's knee-jerk reaction is "that's nonsensical", I suppose, but trying to pin down exactly why it is so sheds some light on what the bases of the PT are usually felt to be. In fact, I do think it might be pedagogically better. You just talk about the outermost shell, and the kids can already see helium is a noble gas anyway because it's the end of the row.
So, in the two contentious cases, the table has passed with flying colours for the group 3 question where we asked for its help, and has shed some new light on the helium situation.
Another analogy between H-Li and He-Be:
| “ | Let us now return to the discussion that Eric Scerri and I had in Kananaskis, Canada a few years back. Can the above presented aspects of the theory of nonadditive effects applied to helium, beryllium and neon cast any light on the question of the “correct” location of He in Mendeleev´s Periodic system? Scerri has devoted some deep philosophical ideas (Scerri 2005) to these matter. We have seen here that the striking similarities between He and Be not only in their atomic spectroscopies, but also in having the two hardest to detect dimers, disappear as soon as nonadditivity is taken into account. By merely taking into account three-body corrections we already see a very large contrast between He and Be, and as concerns the nature of the liquid and solid states of these two elements, all similarities are lost: Be can solidify at room temperature while He is still a gas at temperatures approaching absolute zero. We can say that collective effects are responsible for this, but as we have shown here the dominance of the collective effects for beryllium begins in the Be trimer. In Kananaskis, Eric and I explored the reason for this and concluded that (at least from a physicist’s point of view) it must stem from the Bohr-Schroedinger valence electron shells. Both He and Be have a closed s-subshell, 1s for the former and 2s for the latter. So two Be atoms (as much as would two He atoms among themselves) can effectively interact by resonating through their identical energies. A third Be atom (or in turn three He atoms) cannot resonate effectively because the third Be, by Pauli’s principle needs to approach in an excited state. However in Be the nearest excited state is 2p, marginally higher than 2s, and thus it may still resonate and form a stable trimer (more stable than the dimer at any rate). Contrast the He trimer: the third partner, by Pauli’s principle must also go to the nearest excited state...but this is the much higher lying 2s. Resonance is impossible and He trimers, tetramers and so on up to the infinite solid are no more stable than the dimer. For reasons of space we will not discuss the contributions of nonadditivity in He solvent effects as concerns superfluidity, which is a different story (Blaisten et al. 1979). The net effect of this difference in the separations between the ground and first excited states of He and Be lead to the enormous three and four-body energies of the latter. We may recall the similar situation for the first column of the table where H lies above Li –see however Scerri’s alternative perspective on this (Scerri 2005), where the Li–Li interaction resembles that for H–H molecule. Li atoms however easily resonate, forming larger molecules (del Conde et al. 1981); while H cannot form a stable 3-atom molecule, again due to the great energy difference between 1s and 2s as compared with the small energy separation between 2s and 2p which actually allows the resonance not only of 3 Li atoms but up to eight (of course the partial occupation of these “energy bands” typical of metals is another story). In short we have seen that as concerns the pure pair interactions the similitudes of H and He to Li and Be are large, but the nonadditive corrections (also named few-body or collective effects) to the energies of the beryllium and lithium solids makes for their enormously different physical states as solids. | ” |
| — Octavio Novaro, On the rightful place for He within the periodic table, 2007 | ||
So you see, it is what I said: the difference is the lack of a 1p subshell. Just like a difference between Mg and Ca is the absence of a 2d subshell.
And even from an article critical of He-Be:
| “ | ...one recalls that the leading elements (Li, Be, B, C, N, O and F) show a certain extent of ‘exotic behaviour’, when compared with those below them in the group. The diagonal relationships in the PT (Rayner-Canham 2011), that is similarities between Li and Mg; Be and Al; B and Si, and to a lesser extent of C and P, have been well known for a long time. These similarities might be, at least in part, related to the fact that atoms of the elements of the second period do not have (empty) d-orbitals of energy comparable to that of the valence electrons, unlike their heavier analogues. A similar, but much more pronounced ‘exotic behaviour’ could then be a priori expected for hydrogen, being a first period element (and thus having no energetically close p-orbitals). | ” |
Vladimir M. Petruševski & Julijana Cvetković are here saying that floating H is not so bad, but I beg to differ. This exotic behaviour of H-Li is completely analogous with those of B-Al, C-Si, N-P, O-S, F-Cl, Ne-Ar and vindicates the typical placement of hydrogen over lithium. Having H-Li be that much more extreme establishes the trend: the leading f-elements are not so exotic (but still a bit) compared to the heavier ones, the leading d-elements more so, the leading p-elements significantly more so. Why should not the leading s-elements be significantly more so still? He-Ne shows either no exotic behaviour or the wrong exotic behaviour (lower electronegativity for helium!); He-Be is completely right and requires no breaking apart of blocks.
As Henry Bent said: "Helium above beryllium brings out the best in the Periodic System." Generalities, generalities, generalities, it is all generalities.
At this point, I also note that the first seven periods of the above table are the same as Fig. 8 of a 2017 article by Mikhail Kurushkin (10.1021/acs.jchemed.7b00242).
Extension
- Neither am I with this
Now, how does our criterion fare for the unknown elements?
Period 8
Why does the table have E157-E166 as the 7d elements and not E155-E164 like Fricke and Pyykkö's famous tables basically do? Well, most of all because of the reasonings in the archives of Talk:Extended periodic table: E164 as 7d9s is most analogous to Pd as 4d5s. Or, speaking more usefully about fuzzy configurations, they both have ten valence electrons and have ready access not only to 7d/4d but also to 9s+9p1/2+8p3/2 / 5s+5p respectively. It's interesting that Fricke's original paper was rather equivocal about the groups of those elements, and chemically speaking these are more accurate based on the computations.
[In the following I am mostly using the old CAS-style A/B notation, in which the B is used for the d-block groups, to be consistent with the source. The source does use "VIII" and "0" instead of "VIIIB" and "VIIIA", though. If you have a problem with it, the concordance is as follows, now also including my preferred group labels:
| IA | IIA | IIIB | IVB | VB | VIB | VIIB | VIII | IB | IIB | IIIA | IVA | VA | VIA | VIIA | 0 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 |
| Is | IIs | IIId | IVd | Vd | VId | VIId | VIIId | IXd | Xd | XId | XIId | IIIp | IVp | Vp | VIp | VIIp | VIIIp |
In particular VIII refers to three columns.]
From the paper in Actinides Reviews, 1 (1971) 433–485 (my commentary in brackets):
"Even for the neutral atom, a similarity can be seen between Pd where ten d electrons and no s electrons form the outer shell and element Z = 164." (We might add that the state of affairs in the 7d row, where at most only one electron is promoted to the 9s orbital, and often none are, is like an extended version of the trend that we can see happening in the 4d row, if relativistic effects had not intervened for the 5d and 6d rows.) "Pennemann et al. ... agree with Fricke et al. that the metallic form might be quite stable but they compare it more with Hg whereas Fricke et al predict E164 to be a noble metal which should be in the same chemical group as Pd and Pt." (This placement naturally fixes the position of the preceding d-elements.)
"Here the trend becomes very obvious that the radii and ionization energies of alkaline and alkaline earth elements increase with Z whereas in the first part of the Periodic System they decrease. From this side E165 and E166 will be members of the groups Ia and IIa. From a more chemical point of view, they will be likely more members of the Ib and IIb groups because of the 7d shell which is more comparable to the elements Au and Hg (but also to the elements E119 and E120) as can be seen from Fig. 14. Therefore, higher oxidation states than 1 and 2 might readily occur." (It also seems to me that the placement of E165 and E166 in groups IA and IIA is a symptom of normalising relativistic effects and forgetting how things work in the normal part of the periodic system that we usually deal with, as I noted two sections ago. Not only is the 7d < 9s situation comparable to the normal 3d < 4s, 4d < 5s, and 5d < 6s situations rather than the odd 6d > 7s situation, but also one of the key points of chemistry is that there is a large energy gap between a closing p-shell and the s-shell of the next principal quantum number, so you cannot take electrons out of an earlier shell. We accept it, perhaps, for E119 and E120 because we then have no choice, but since E157–E164 and E167–E172 return to a non-relativistic-like situation, it makes sense to draw d-block analogies for E165 and E166 – especially since here we again have the group IB- and IIB-like situation with a weaker shielding provided by the d-subshell, and if the gap is bigger between 7d and 9s than it was for the other d-series, well, that follows the trend that we see incipient in the 4d series. Thus it is reassuring to see that E165 and E166 continue the trend of falling ionisation energies for the non-relativistic Cu-Ag and Zn-Cd series, which had suffered an interruption at the relativistic Au-Rg and Hg-Cn.)
Note that their table 10 of predicted properties gives as the most analogous group for E157–E164 the expected IIIB through VIII, and for E167–E172 the expected IIIA through 0; we follow these, only squeezing E165 and E166 instead into the gap of IB and IIB instead of IA and IIA.
From the 1975 paper:
Again, the same table with the group assignments is given.
"In the periods before the 8th period, normally all d and p elements are influenced in their chemical behavior more or less by the outer s electrons. This is no longer true for the d transition elements 155 to 164, where the 8s and 8p1/2 eleetrons are bound so strongly that they do not participate in the chemical bonding. Fig. 22 shows the outer electronic wave functions of element 164 with the deeply buried 8s and 8p1/2 electrons. This electronic structure is quite similar to that of the d elements of the lower periods, where the outer s electrons are removed." (Well, in many chemical environments the configuration of such a d-element is indeed ds! And let's not forget what I said earlier about this continuing the trend towards disfavouring s-occupancy even in the gaseous atom that we already see in the 4d series.) One might therefore argue that, as a first guess, the aqueous and ionic behavior of an E ion of the lower d elements is comparable to an E ion of elements 155 to 164 after making allowance for the different ionic sizes and charge. But because the 9s and 9p1/2 states are easily available in 164 for hybridization, the chemical behavior is expected not to be too different from that of the other d elements." (Therefore we see that we must mentally think of 9s rather than 8s as the covering s-shell here. And indeed we sometimes see promotions to there in the ground state: compare the 4d and 7d series! I here use the predictions in the table at the bottom of this article:)
| 4d | Y | Zr | Nb | Mo | Tc | Ru | Rh | Pd | Ag | Cd |
|---|---|---|---|---|---|---|---|---|---|---|
| 4d5s | 4d5s | 4d5s | 4d5s | 4d5s | 4d5s | 4d5s | 4d5s | 4d5s | 4d5s | |
| 7d | 157 | 158 | 159 | 160 | 161 | 162 | 163 | 164 | 165 | 166 |
| 7d9s | 7d9s | 7d9s | 7d9s | 7d9s | 7d9s | 7d9s | 7d9s | 7d9s | 7d9s |
Eccola! And if you will see in some reputable places slightly different predictions with regard to which are 9s and 9s, well, that just goes to show that 7d and 9s are nearly degenerate, doesn't it? Surely it is nice to see such nearly exact homology, though! ^_^)
(This consideration is why I disagree with Fricke's argument for his placement of E165 and E166, which goes as follows:)
"From the normal continuation of the periodic table one would expect that after the completion of a d shell (at element 164) two elements in the IB and IIB chemical groups should appear. In a very formal way this is true, because with the filling of the 9s electrons in elements 165 and 166 there are outer s electrons chemically available. On the other hand, these outer s electrons should be the ones which began with the onset of the period. The 8s electrons are already very strongly bound so that the two 9s electrons which are filled in have to be assumed to define the beginning of a new period." (But as we can see, already by the time we get out of the quagmire of superactinides, it is 7d and 9s which are running the show, as if 7d was a transition series in between 9s and the hybrid 9p1/2+8p3/2 that are nearly degenerate and act like the 3p shell. So there is a slow transition between thinking of 8s as the outer s-shell and replacing it with 9s. Now, their comparisons of ionisation energies and atomic radii have some force. So let's draw a table:)
| Li | Na | K | Rb | Cs | Fr | 119 | 165 |
|---|---|---|---|---|---|---|---|
| 152 | 186 | 227 | 248 | 265 | (~255?) | (240) | (210) |
| Cu | Ag | Au | Rg | 165 | |||
| 128 | 144 | 144 | (152 or 138) | (210) | |||
| Be | Mg | Ca | Sr | Ba | Ra | 120 | 166 |
| 112 | 160 | 197 | 215 | 222 | (~225?) | (200) | (180) |
| Zn | Cd | Hg | Cn | 166 | |||
| 134 | 151 | 151 | (160 or 147) | (180) |
(Metallic radii are in picometres for the stable elements from Atomic radii of the elements (data page), and from Fricke for Rg, Cn, and the undiscovered ones. Figures for francium and radium are shameless WP:OR based on the covalent radius supplemented by graphomancy from Fricke's Fig. 10 in this paper, suggesting Fr is around the average of those of Rb and Cs, and Ra is a bit larger than Ba, but they are not the point anyway *handwaves*.)
(Now let's show ionisation energies, because if you've read till here you probably want me to:)
| Li | Na | K | Rb | Cs | Fr | 119 | 165 |
|---|---|---|---|---|---|---|---|
| 520.2 | 495.8 | 418.8 | 403.0 | 375.7 | 380 | (462.0) | (520) |
| Cu | Ag | Au | Rg | 165 | |||
| 745.4 | 731.0 | 890.1 | (1020) | (520) | |||
| Be | Mg | Ca | Sr | Ba | Ra | 120 | 166 |
| 899.5 | 737.7 | 589.8 | 549.5 | 502.9 | 509.3 | (563.3) | (630) |
| Zn | Cd | Hg | Cn | 166 | |||
| 906.4 | 867.8 | 1007.1 | (1155) | (630) |
(Data from molar ionisation energies of the elements and this page. Neither trend looks all that great, as both demand some sort of about-face; but if we consider Au–E120 part of the "relativistic effects zone" that should be exceptional, against the trend of the other elements, it seems to me that putting E165 and E166 in groups 11 and 12 respectively makes more sense as then the trend goes back to normal once we go into the "pretending to be out of the relativistic effects zone" that includes E157–E173. If we can stomach E119 and E120 as A-group elements with B-group tendencies, then surely we can also stomach E165 and E166 as B-group elements with A-group tendencies. The much lower density of E165 and E166 compared to E156–E164 can to some extent be considered an illustration of this.)
TL;DR: the classification of E157–E164 as group 3–10 elements is supported by Nefedov et al. in their periodic table illustration, by Droog Andrey's popularisation article, and also by Fricke's table in his papers giving assignments of what he calls groups IIIB through VIII; Fricke himself admits that this is the most chemically relevant classification with E164 as a heavy congener of Pd and Pt. The classification of E165 and E166 as B-subgroup rather than A-subgroup elements is at least partially supported by Fricke's papers as well, and is the classification used by Droog Andrey (like E119 and E120, they do mix characteristics of A-subgroup and B-subgroup elements); finally E167–E172 present no problems (well, apart from us drawing them in period 8 rather than 9, but that is obvious and also what his article does.)
So we see we have a chemically sensible 8th row classification that leads to a period of strong homology between the 4d/5p and 7d/8p elements.
Period 9
E173, with its single 6g valence electron, is probably an illustration of a "delayed collapse" of 6g that has become a premature collapse thanks to the 9s and 9p1/2 shells preemptively going into period 8 (like intruder levels from nuclear shells).
Judging from E184 we seem to have a situation of 6g7f8d, without an outer covering 10s+10p1/2(?) subshell. Maybe that will come later, but this is kind of like what relativistic destabilisation of high-angular-momentum subshells gives us already in 6d and 7d, just even more so, right? And it's pretty much like the very weak covering of 5s over 4d in the 4d metals, so all is well. I would expect E173-E178 to be something like Fr through U as a trend if relativistic effects hadn't spoilt our fun, and then a sort of "uranide" series judging from Fricke et al.'s predictions of E184 (which match eka-E130). Maybe the bonding early in this series will show more 6g involvement and provide us the chance for 50-electron rule. ;) When this series should end, I cannot say, but maybe we can rationalise it as (6g7f8d10s10p1/2). BTW, that is almost exactly like what we see from E119 onwards...perhaps we may yet get a real homology if 11s and 11p1/2 drop in and thence a noble gas at E226? Well, I have drawn that as a guess putting faith in periodicity. If it happens, periodicity wins; and if it doesn't happen, I'm eager to learn what does happen instead!
Theorists may wish to get their computers running to chart period 9, since an island of stability may appear around E210 (perhaps dvi-nobelium = tri-ytterbium if the analogy holds up with 6g7f8d10s10p1/2 ~ 5g6f7d8s8p1/2?), although this is very much a relative thing since the expected half-life is on the order of microseconds. (For the one predicted at E274, the expected half-life is too short to even think about doing any chemistry, being on the order of zeptoseconds.) ^_^ The reason? Having a period 9 to confirm or disprove this placement of period 8 would be an invaluable test. If periodicity is really holding the fort qualitatively even through the storm of relativistic effects, I would guess a period 9 that really shows true homology to period 8, and I want to know if this is likely or not. We'll have to accept likely not having a period 10, though. No h block elements! :(
What is a metal?
I used to worry about this, but I now think it doesn't completely make sense here. The periodic table is essentially a classification of atoms and elements, not of simple substances. Metallicity however does focus on simple substances, as explicit in the solid-state physics idea of metallicity, which is about band structure. Well, it's just like getting a bunch of atoms together, their orbitals mix just like two hydrogens forming a H2 molecule, you get a wide valence band for a metal. So it is really based on electronic structure, and translates to results about the physical substance. But that is not different between pure Na metal and a NaK alloy, say. Just like covalent bonding is not different whether it is H2 or HF. In either case you get things about the substance, and something that makes sense for non-simple substances is not really an intrinsic elemental property. Just like melting point, reactivity, etc.
Of course it also correlates imperfectly but somewhat interestingly well with the chemical properties, but that is because both come from the electronic structure. High atomic radius, low ionisation energies, these will help physical metallicity for electron delocalisation just as it helps chemical metallicity. But none of the chemical properties are at all intrinsic to what exactly a metal is, which is defined by the delocalisation, the band structure, the bonding type. The simple cation or basic oxides has nothing to do with it in fact, it is all about oxidation state and chemical affinity to different ligands; that's why you can get things like tungsten which are physically superb metals but show neither, and germanium which is physically totally not a metal but forms a simple Ge cation in aqueous solution anyway (10.1002/jcc.21315, it's just not so emphasised because germanium is more stable in the +4 state). It is not about metallicity but rather about other things you can get from periodicity, let's not conflate the two.
Therefore I say, leave the definition of what a metal is to the solid state physicists! And just note that chemically speaking it may differ. DA has suggested some good definitions, such as one based on Fermi surfaces, and one based on bonds to neighbours. But I prefer to just colour blocks as a classification of atoms and elements. This distinction between a classification of elements and a classification of simple substances is also why I think helium over beryllium is perfectly fine.
Usual categories
Anyway the usual classifications are a bit silly IMHO. Well, group 12 has weak transition behaviour. So does group 3. Meanwhile so do heavy group 4 and group 5. Actually it is just general trends, edge groups have properties of their neighbours. Be and Mg are not alkaline, so why should they be called alkaline earth metals? There's just a misguided idea that categories have to match groups, it isn't necessarily so. Maybe this is why some people are freaked out by hydrogen in group 1; do they think that that means you have to call it an alkali metal?
I believe: categories do not have to match the group. Therefore I prefer "restricted families". Icosagen for group 13 is cute but only really works well for boron. ;)
- Alkali metals – Li, Na, K, Rb, Cs, Fr
- Alkaline earth metals – Ca, Sr, Ba, Ra
- Crystallogens – C, Si, Ge (Sn is ambiguous, Pb is not a good inclusion but maybe for comparison)
- Pnictogens – N, Si, As (Sb is ambiguous, Bi only for comparison)
- Chalcogens – O, S, Se, Te (Po is ambiguous)
- Halogens – F, Cl, Br, I (At is ambiguous)
- Noble gases – He, Ne, Ar, Kr, Xe, Rn (split across groups, yes)
But the weirdest of all is "lanthanide". The old definition excluding La was the sillier one (just look at how much you want to include La for comparison anyway). Well, now we have the same problem but with yttrium. ;)
Now, simply put:
- Lu = 4f 5d 6s, has three electrons outside a stable 4f core, almost always uses all three to form a stable Lu oxidation state, with similar radius and position on the acidic-basic continuum to its lighter congener Y due to poor shielding by the 4f core electrons. The two (Y and Lu) are almost invariably found together.
- Hf = 4f 5d 6s, has four electrons outside a stable 4f core, almost always uses all four to form a stable Hf oxidation state, with similar radius and position on the acidic-basic continuum to its lighter congener Zr due to poor shielding by the 4f core electrons. The two (Zr and Hf) are almost invariably found together.
- Ta = 4f 5d 6s, has five electrons outside a stable 4f core, almost always uses all five to form a stable Ta oxidation state, with similar radius and position on the acidic-basic continuum to its lighter congener Nb due to poor shielding by the 4f core electrons. The two (Nb and Ta) are almost invariably found together.
Compare:
- La = 5d 6s, has three electrons outside a stable core, almost always uses all three to form a stable La oxidation state, but due to the hard noble gas core is very much larger and more basic than its purported lighter congener Y. Indeed they are often not so closely associated as Lu and Y, because the early and late lanthanides separate from each other, and Y patterns as a late lanthanide due to its size. Not to mention that it often uses its 4f orbitals to bond in complexes, which the other 5d elements never do.
So, who is creating a great anomaly within 5d? Whereas:
- Yb = 4f 6s, has 4f as a valence orbital, and the core is only . Sometimes uses only the 6s electrons, sometimes throws in a 4f one, and is close to the border between divalence and trivalence in the metallic state (but on the divalent side). No clear lighter congener.
- Tm = 4f 6s, has 4f as a valence orbital, and the core is only . Sometimes uses only the 6s electrons, sometimes throws in a 4f one, and is close to the border between divalence and trivalence in the metallic state (but on the trivalent side). No clear lighter congener.
See the graph for divalent and trivalent metallic states. The trend towards +2 at the end of the Ln and An, followed by going straight back to +3 for Lu and Lr, is exactly like the 3d trend of Mn-Zn favouring +2 mostly followed by Ga back to +3.
So, it seems absolutely clear that the trend in the Ln is really ending at Yb, and that Lu is something different that patterns totally with Hf through Hg?
No one loses sleep that Sc and Y are not lanthanides, and the difference of blocks is important here, because the absence of 4f vs. 4f interplay for Lu makes some difference. For Lr it is even more clear-cut, as Lr +3 totally breaks the trend towards +2 in the late An (second-row inner TMs).
Why the rectangles?
Firstly, I will note that because the strong symmetry break is clearly between a noble gas (well, treating Og as one) and the next alkali metal—the only place where all important trends break—I think it is not good to do a cylindrical periodic table or anything that doesn't make this break clear.
In my opinion, the valence electrons are important enough that the rectangular form that emphasises them ought to be the ideal one. The Bayley-Zmaczyński-Thomsen-Bohr pyramid may be a "next-best" option for showing the secondary periodicities, but I think it's more important to keep the block labels there as simple as possible.
Electronegativity
While Wulfsberg's approach is excellent, I prefer using chemically sounder electronegativity values than Pauling (that is awkward for elements like Mo and W that favour cluster formation)! (The table is transcluded from User:Double sharp/Electronegativity.)
Electronegativity scale for all 118 known elements from this table by A. V. Kulsha and T. A. Kolevich.
{kind=link}
Link for my own reference: where it perhaps started.
| Z | Element | χ |
|---|---|---|
| 1 | hydrogen | 2.20 |
| 2 | helium | 3.20 |
| 3 | lithium | 1.00 |
| 4 | beryllium | 1.50 |
| 5 | boron | 2.00 |
| 6 | carbon | 2.50 |
| 7 | nitrogen | 3.00 |
| 8 | oxygen | 3.50 |
| 9 | fluorine | 4.00 |
| 10 | neon | 4.50 |
| 11 | sodium | 0.90 |
| 12 | magnesium | 1.30 |
| 13 | aluminium | 1.70 |
| 14 | silicon | 1.90 |
| 15 | phosphorus | 2.24 |
| 16 | sulfur | 2.64 |
| 17 | chlorine | 3.06 |
| 18 | argon | 2.94 |
| 19 | potassium | 0.80 |
| 20 | calcium | 1.10 |
| 21 | scandium | 1.33 |
| 22 | titanium | 1.40 |
| 23 | vanadium | 1.48 |
| 24 | chromium | 1.56 |
| 25 | manganese | 1.52 |
| 26 | iron | 1.60 |
| 27 | cobalt | 1.64 |
| 28 | nickel | 1.69 |
| 29 | copper | 1.77 |
| 30 | zinc | 1.71 |
| 31 | gallium | 1.80 |
| 32 | germanium | 1.96 |
| 33 | arsenic | 2.22 |
| 34 | selenium | 2.52 |
| 35 | bromine | 2.86 |
| 36 | krypton | 2.70 |
| 37 | rubidium | 0.77 |
| 38 | strontium | 1.05 |
| 39 | yttrium | 1.28 |
| 40 | zirconium | 1.35 |
| 41 | niobium | 1.44 |
| 42 | molybdenum | 1.53 |
| 43 | technetium | 1.51 |
| 44 | ruthenium | 1.62 |
| 45 | rhodium | 1.68 |
| 46 | palladium | 1.73 |
| 47 | silver | 1.79 |
| 48 | cadmium | 1.66 |
| 49 | indium | 1.74 |
| 50 | tin | 1.86 |
| 51 | antimony | 2.04 |
| 52 | tellurium | 2.28 |
| 53 | iodine | 2.58 |
| 54 | xenon | 2.39 |
| 55 | caesium | 0.70 |
| 56 | barium | 0.92 |
| 57 | lanthanum | 1.11 |
| 58 | cerium | 1.13 |
| 59 | praseodymium | 1.14 |
| 60 | neodymium | 1.15 |
| 61 | promethium | 1.16 |
| 62 | samarium | 1.17 |
| 63 | europium | 1.09 |
| 64 | gadolinium | 1.20 |
| 65 | terbium | 1.21 |
| 66 | dysprosium | 1.23 |
| 67 | holmium | 1.24 |
| 68 | erbium | 1.25 |
| 69 | thulium | 1.26 |
| 70 | ytterbium | 1.19 |
| 71 | lutetium | 1.31 |
| 72 | hafnium | 1.38 |
| 73 | tantalum | 1.46 |
| 74 | tungsten | 1.54 |
| 75 | rhenium | 1.55 |
| 76 | osmium | 1.67 |
| 77 | iridium | 1.75 |
| 78 | platinum | 1.84 |
| 79 | gold | 1.93 |
| 80 | mercury | 1.81 |
| 81 | thallium | 1.78 |
| 82 | lead | 1.82 |
| 83 | bismuth | 1.88 |
| 84 | polonium | 1.98 |
| 85 | astatine | 2.09 |
| 86 | radon | 1.94 |
| 87 | francium | 0.72 |
| 88 | radium | 0.85 |
| 89 | actinium | 0.97 |
| 90 | thorium | 1.01 |
| 91 | protactinium | 1.04 |
| 92 | uranium | 1.06 |
| 93 | neptunium | 1.08 |
| 94 | plutonium | 1.12 |
| 95 | americium | 1.07 |
| 96 | curium | 1.18 |
| 97 | berkelium | 1.22 |
| 98 | californium | 1.27 |
| 99 | einsteinium | 1.32 |
| 100 | fermium | 1.36 |
| 101 | mendelevium | 1.39 |
| 102 | nobelium | 1.37 |
| 103 | lawrencium | 1.29 |
| 104 | rutherfordium | 1.34 |
| 105 | dubnium | 1.41 |
| 106 | seaborgium | 1.49 |
| 107 | bohrium | 1.59 |
| 108 | hassium | 1.72 |
| 109 | meitnerium | 1.83 |
| 110 | darmstadtium | 1.92 |
| 111 | roentgenium | 1.99 |
| 112 | copernicium | 1.91 |
| 113 | nihonium | 1.87 |
| 114 | flerovium | 1.85 |
| 115 | moscovium | 1.57 |
| 116 | livermorium | 1.65 |
| 117 | tennessine | 1.76 |
| 118 | oganesson | 1.61 |
Or in tabular form, with a pretty colour gradient:
| Is | IIs | IIIf | IVf | Vf | VIf | VIIf | VIIIf | IXf | Xf | XIf | XIIf | XIIIf | XIVf | XVf | XVIf | IIId | IVd | Vd | VId | VIId | VIIId | IXd | Xd | XId | XIId | IIIp | IVp | Vp | VIp | VIIp | VIIIp |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H 2.20 |
He 3.20 |
||||||||||||||||||||||||||||||
| Li 1.00 |
Be 1.50 |
B 2.00 |
C 2.50 |
N 3.00 |
O 3.50 |
F 4.00 |
Ne 4.50 | ||||||||||||||||||||||||
| Na 0.90 |
Mg 1.30 |
Al 1.70 |
Si 1.90 |
P 2.24 |
S 2.64 |
Cl 3.06 |
Ar 2.94 | ||||||||||||||||||||||||
| K 0.80 |
Ca 1.10 |
Sc 1.33 |
Ti 1.40 |
V 1.48 |
Cr 1.56 |
Mn 1.52 |
Fe 1.60 |
Co 1.64 |
Ni 1.69 |
Cu 1.77 |
Zn 1.71 |
Ga 1.80 |
Ge 1.96 |
As 2.22 |
Se 2.52 |
Br 2.86 |
Kr 2.70 | ||||||||||||||
| Rb 0.77 |
Sr 1.05 |
Y 1.28 |
Zr 1.35 |
Nb 1.44 |
Mo 1.53 |
Tc 1.51 |
Ru 1.62 |
Rh 1.68 |
Pd 1.73 |
Ag 1.79 |
Cd 1.66 |
In 1.74 |
Sn 1.86 |
Sb 2.04 |
Te 2.28 |
I 2.58 |
Xe 2.39 | ||||||||||||||
| Cs 0.70 |
Ba 0.92 |
La 1.11 |
Ce 1.13 |
Pr 1.14 |
Nd 1.15 |
Pm 1.16 |
Sm 1.17 |
Eu 1.09 |
Gd 1.20 |
Tb 1.21 |
Dy 1.23 |
Ho 1.24 |
Er 1.25 |
Tm 1.26 |
Yb 1.19 |
Lu 1.31 |
Hf 1.38 |
Ta 1.46 |
W 1.54 |
Re 1.55 |
Os 1.67 |
Ir 1.75 |
Pt 1.84 |
Au 1.93 |
Hg 1.81 |
Tl 1.78 |
Pb 1.82 |
Bi 1.88 |
Po 1.98 |
At 2.09 |
Rn 1.94 |
| Fr 0.72 |
Ra 0.85 |
Ac 0.97 |
Th 1.01 |
Pa 1.04 |
U 1.06 |
Np 1.08 |
Pu 1.12 |
Am 1.07 |
Cm 1.18 |
Bk 1.22 |
Cf 1.27 |
Es 1.32 |
Fm 1.36 |
Md 1.39 |
No 1.37 |
Lr 1.29 |
Rf 1.34 |
Db 1.41 |
Sg 1.49 |
Bh 1.59 |
Hs 1.72 |
Mt 1.83 |
Ds 1.92 |
Rg 1.99 |
Cn 1.91 |
Nh 1.87 |
Fl 1.85 |
Mc 1.57 |
Lv 1.65 |
Ts 1.76 |
Og 1.61 |
You can use electronegativity along with cation charge and radius to predict the degree of hydrolysis of cations (again, this is Fajans).
| OS | Basic | Amphoteric | Acidic |
|---|---|---|---|
| +1 | Li, Na, K, Cu, Rb, Ag, In, Cs, Hg, Tl, Fr, Mc | H, I, At, Nh | Cl, Br |
| +2 | Mg, Ca, Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Sr, Mo, Tc, Ru, Rh, Pd, Ag, Cd, Ba, Sm, Eu, Yb, Pt, Hg, Po, Ra, Fm, Md, No, Ds, Cn, Lv | Be, Zn, Ge, Sn, Pb, Fl, Og | |
| +3 | Ti, V, Mn, Y, Mo, Ru, La, Ce, Pr, Nd, Pm, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb, Lu, Re, Ac, U, Np, Pu, Am, Cm, Bk, Cf, Es, Fm, Md, No, Lr, Mt | Al, Sc, Cr, Fe, Co, Ni, Cu, Ga, Rh, Ag, In, Sb, Ir, Au, Tl, Bi, At, Rg, Mc, Ts | B, C, N, P, S, Cl, As, Br |
| +4 | Th, Pa, U, Np, Pu, Am, Rf | Ti, V, Cr, Mn, Fe, Co, Zr, Nb, Mo, Tc, Ru, Rh, Pd, Sn, Ce, Pr, Tb, Hf, Ta, W, Re, Os, Ir, Pt, Pb, Po, Cm, Bk, Hs, Mt, Ds, Lv, Og | C, N, Si, S, Cl, Ge, Se, Te |
| +5 | Pa, U | V, Nb, Ta, Np, Pu, Am, Db | N, P, S, Cl, Cr, Mn, Fe, Co, As, Br, Mo, Tc, Ru, Sb, I, W, Bi, At |
| +6 | U, Np, Pu, Am, Cm, Sg | S, Cr, Mn, Fe, Se, Mo, Tc, Ru, Te, Xe, W, Re, Os, Po, Rn, Bh, Hs | |
| +7 | Np, Pu | Cl, Mn, Br, Tc, Ru, I, Re, Os, At, Bh | |
| +8 | Ru, Xe, Os, Hs |
Basically, it is charge squared over ionic radius with a correction for electronegativity.
Lesson plan
This is how I'd see a lesson go once we finally manage to dump the historical relic that is the La table:
- The most important thing to understand is the nature of the blocks and the subshells (I trust we already know what n and the shells are). We know of four now (s, p, d, and f), and almost certainly within the very near future we will open a fifth (g). These are distinguished by a certain quantity called l: s, p, d, f, and g correspond to l values of 0, 1, 2, 3, and 4 respectively. The names s, p, d, and f come from spectroscopy, before we really understood the regularity. Now that we do know it, future ones go alphabetically (whence g).
- We may look at their shapes, how many of each there are, how far they extend from the nucleus, and the important concept of radial nodes. You don't need to understand what these mean now, because there is some complicated mathematics here: the important thing is that the first subshell of any type is smaller than you'd expect and often acts rather exceptionally.
- Most tables you encounter will show ground-state electron configurations in the gas phase, which are OK for the s and p elements as an understanding. Those are the elements where you begin.
- In chemically bound environments, the d and f elements switch between many configurations of about equal energy level, and our best scientists have predicted the same for the g elements. The ground-state electron configurations for these elements do not really mean anything much for chemistry for this reason, and you will therefore not be tested on those exceptional configurations even when we do get to basic d block chemistry. Unless you do spectroscopy or molecular gas-phase reactions you'll hardly ever need to know them.
- The important invariant is valence subshells and how many electrons are held within them, in which the PT displays an astonishingly perfect regularity. The better understanding here comes from "fuzzy configurations", e.g. La = (4f5d6s6p), Lu = 4f (5d6s6p). What this means is that we write the core first (the electrons not participating), and then write how many electrons are participating and what subshells they happen to come from. Although we can distinguish which ones are being filled up at any given Z, and call it the characteristic subshell that defines the block, we have to look at all of them as they are being used together and can mix for hybridisation.
- There are important heuristic ideas of what configurations are important for the d and f elements based on important motifs in the PT. For example, in the d elements of groups 3 through 10, it is best to assume a d configuration in compounds. Also for example, the important thing we see in the f elements is the "reserve area" nature of the deeply buried and small f orbitals, where electrons may be promoted from into bonding MOs: this is one way in which we see that La-Yb are real f block elements and Lu is something different. You don't need to know all of this now: if you go further into d and f block chemistry you will learn it then. And maybe when you grow up you will become the pioneers of g block chemistry! Now that you know something about what those orbitals are like, you may not be surprised to here that it is expected to be something like f block chemistry, but also starting an unprecedented situation that builds on it.
- Blocks are delineated by which subshells are valent. For most elements, we will have many subshells active. The order of energy of subshells generally follows Madelung's n+l rule: orbitals fill in order of increasing n+l, and within each group of equal n+l, in order of increasing n. (Also consider Hund's rule here, which is important for light elements.) With the exception that the important gap comes before each s-orbital (not between values of n+l), which therefore fraternises with the "wrong" company. We will see more of this exception soon.
- The Madelung rule is thus restored to perfection. It gives the characteristic "two rows at a time" structure of the periodic table, and something called "double periodicity" that you see across each group: "kinks" in the trend occur every time a block is inserted. Except for the s block; we'll get back to that in a moment.
- We will test you on using the Madelung rule throughout the table, even for d and f elements. For those elements you may either write a "fuzzy configuration" or simply assume everything holds as the Madelung rule expects, e.g. La = 4f6s. If you go for the second option, you are expected to include a disclaimer for those elements that you know that that might not be the ground state but that it will be close and it will be chemically relevant.
- We notice that elements tend to use subsets of each group of orbitals: if we look across row 6, we see the s block (we'll come back to it in a moment). Then we see the f block, where 4f, 5d, 6s, and 6p are all being used: all four in one group. Then the d block, where 4f stops being used, and only the other three are left. Then the p block, where 5d stops being used, and only the last two are left. The characteristic subshell is the one of highest l that is active, and we use it to name the block. Each corresponds to filling its characteristic subshell from empty to full.
- The weirdo is the s block. Nominally, like the others, it corresponds to filling its characteristic subshell. But it has no qualms about using ones of higher l than it. The only really pure s elements are H and He: everybody else has no problem using p or even d orbitals. Not only that, but we can see that due to peculiar gap movement in the Madelung rule (in a sense, a symmetry break from n+l), it can never show any double periodicity: H and He form a huge exception, then 1s shields perfectly, but after that there's never anything cutting in line between a p-subshell and the next s-subshell. We can see this straight from the pattern of the periodic table.
- The duet, octet, 18-, and 32-electron rules (the last mostly unattainable because of buried f-electrons, except for early actinides) come from an element's drive to "fill up" all of these vacancies in its valence subshells.
- We also see the important rule of subshell energy level (e.g. 4f < 5d < 6s < 6p) which goes in order of increasing n within each group, with the understanding that the s-block constitutes an important exception where the s subshell cuts the queue to fill first. In order to emphasise the different regions in which each of those rules are applicable, we draw the blocks absolutely distinct and separate, even with helium over beryllium.
- Most people will not draw He over Be, but over Ne. This is because He is a noble gas in physical and chemical properties: its only shell is full, so it doesn't desire anything to change that status, which makes it quite like Ne and its congeners. However, the block difference makes a big impact for atomic properties. Moreover, H and He then stand as the first-row anomaly for the s block just like the other ones if we put them over Li and Be, which creates a deeper regularity. You will just have to remember that although H and He are all the way on the left side of our table, they are nonmetals. It will turn out that the position means something: H has many metal-like properties in its chemistry, and when our top theoreticians investigated what happens when He is forced into bonding, it seems to want to behave more like Be than Ne.
- The Madelung rule allows one to predict these "fuzzy configurations" absolutely perfectly. If you wish, you may consider filling up the exact configuration according to it: while this will not always be right for the ground state, what you come up with will be chemically relevant, and you can often use it with some heuristics that you'll learn to predict how many electrons will be used for chemistry.
- Some old tables put La and Ac into the d block instead of Lu and Lr. You will often see this if you read some old books (many of which are still quite good when it comes to describing the chemistry), but we will not use this classification. The historical reason for this is that we previously did not know about the f block: the 5f elements that we knew "pretended" to be d elements (we will explain why this happens at a much later date). Therefore we thought all fifteen lanthanides belonged below yttrium, and we misguidedly picked La as the prototype. This mistake obscures the point of blocks, and drags group 3 into the odd lack of double periodicity that should by rights only appear in the s block. It ruins many regularities in the periodic table's trends because they all, at length, lead to this block filling and Madelung rule. It does more harm than good and you shouldn't use it anymore. If you wish, you may do some research and presentations on old periodic tables. You will see really marvellous things from today's perspective, since we only had the chemical properties and we didn't yet know the physics and mathematics that was behind that. You may learn a bit from them about secondary periodicities, and we may even (when dealing with those groups) do a comparison with the secondary linkages; but we have picked the table that is the most consistent and goes for the most important linkages. We respect and honour our past, but we continue building on it, and don't stop there.
- When we get to really heavy elements, like for those g elements shrouded in mystery that maybe some of you will penetrate, but more at home for those elements from the sixth and seventh rows, Einstein's theory of relativity comes into effect. You see it every time you look at a beautiful golden ring, watch the mercury in your thermometer flow (do people see those much anymore?), and see a car running on a lead-acid battery. When you look at those elements later you will only need to understand for now the qualitative aspects, which are in fact mostly about the orbitals. The point is that in each part of the periodic table you must understand how the orbitals have become different, and what that means from the effect of the valence ones on chemistry and physics.
That concludes my tale. Not a single false word has been spoken, but I think it would do some good. And not a word has been spoken about ill-defined and irrelevant differentiating electrons.
There is nothing revolutionary here, everything I say has been said by somebody else who knows more chemistry. ;)
Also, point 14 will need rewriting in the even further future when we dump the placement of He over Ne. ;)
In fact, I did start to write it.
What about the IUPAC survey?
Yeah, I know. It's true that if you count periodic tables in textbooks from the 1970s to the 2010s, as IUPAC did you end up with a 4:1:1 ratio for La:Lu:*. That's an oft-quoted figure in this discussion. The only problem is that this is not telling the full story.
| Decade | La under Y | Lu under Y | * under Y | Total |
|---|---|---|---|---|
| 1970s | 18 69% |
2 8% |
6 23% |
26 |
| 1980s | 31 78% |
6 15% |
3 8% |
40 |
| 1990s | 27 82% |
2 6% |
4 12% |
33 |
| 2000s | 38 62% |
14 23% |
9 15% |
61 |
| 2010s | 16 48% |
6 18% |
11 33% |
33 |
The popularity of La is buoyed by historical data up to the 1990s. Then came WebElements showing Lu in the 2000s, thereby immediately giving this issue much more publicity. Remember what Jensen said in 1982: "Indeed, in talking with his fellow chemists, the author discovered that none of them was aware of the evidence favoring the reassignment of lutetium and lawrencium or indeed that there ever was any question about their placements (a category in which the author must include himself until very recently)."
So: thank you so much, Prof. Winter!
The future?
Do I think that this table will win in the future? (By now you know I think it should win.)
Yes, if we wait long enough. Not that soon though, probably. After all, the inconsistency regarding how the Madelung rule and ionising transition metals are usually thought isn't dead yet, is it? (The one where 4s is said to fill before 3d because it's lower in energy, but also ionise first before 3d because it's also higher in energy.) But it doesn't matter. We know it is right, we can click our tongues in dismay when we inevitably see the worse form. XD
Am I sure that what I say is right? Yes, I've actually checked most of this with experts, both in person and from papers and textbooks. Am I also sure that I will continue to be annoyed that nobody follows it for a good long while? Yes. It's just that here I am more hopeful since the actinides did get shifted half-properly (unfortunately starting from thorium instead of actinium, but at least they got shifted).
Well, IUPAC has a project working on the group 3 issue, started in 2015. That's a bit late since Henry Bassett already knew in 1892 that scandium and yttrium are chemically not close to lanthanum and put them in separate groups, but better late than never. It has both La and Lu proponents on it. We have already addressed all arguments of La proponents to my satisfaction even if not to theirs. ;) So, we have some chance that they will pick the better answer, and some chance that they will not. But that's OK. Thanks to Prof. Mark Winter everyone knows about this dispute by now, everyone can start investigating the merits of them. If they don't pick Lu, then I bet the opposition saying Lu is better will keep on going strong until they have to look at it again. And if they do pick Lu, then soon enough we will have La as a fading memory since once Lu becomes standard, everyone will wonder what the point of introducing a strange gap to group Sc and Y with chemically less similar La is.
Helium over beryllium can maybe wait a little while longer. I'm sure that once it wins people will stop grumbling too, since the grumbling about hydrogen over lithium has mostly dissipated. With a bit of luck we can teach the kainosymmetry and then kids will stop wondering "why is a nonmetal over on the wrong side" and start thinking "oh, the first row of every block is significantly less strongly metallic in character". Just look at 1s vs 2s, 2p vs 3p, even to some extent 3d vs 4d and 4f vs 5f (the latter are metals vs metals but you can compare oxidising power in highest oxidation states). Be wise, generalise, as Gertrude Ehrlich says in her book on abstract algebra. ;)
What about Misplaced Pages?
Well, all the statements from reliable sources are fair game. The problem is that the Lu form itself is not. That's the problem, that the literature reacts to this sort of information with about the activity of argon. It is common enough that mentioning that there is an argument and summarising it is already warranted, but not so common yet as to convince most editors here that it should be changed as the default.
Maybe you can make the argument that the majority of authors who actually think about this support Lu, and that those sources are more important, but then there is one little problem: the majority of authors who actually think about helium's position ask for it to be moved, because most people don't see anything wrong with the current form. And maybe you can make the argument that in fact the majority of authors are in fact saying things that support the Lu arguments and oppose the La arguments, and that writing down all of those and then putting a La table up looks strange, but the only problem is that none of them seem to be actually aware that what they say has any effect on the dispute or that there is a dispute at all. So neither of the arguments really works, for now I cannot do anything to change it on WP. Therefore, I wait for IUPAC.
I do use this form exclusively outside Misplaced Pages. (Yes, with the funny helium.) That is when I am actually thinking about chemistry, which is not that often anymore. In fact, if it were not for the Eka-Yttrium Holy War starting up on Misplaced Pages again in 2019–20, I would not have thought enough about this to complete my idea and be able to write something useful here. So there's that to thank it for even if I think I have gotten everything I wanted about periodicity already. Now I just wait for more predictions to update things like late transactinide metallicity and get better data on periods VIII and most especially the totally guessed IX.
Summary
- Group 3. Lanthanum and actinium have low-lying f orbitals that contribute to chemical bonding, and lutetium and lawrencium do not. Also chemically and physically speaking Sc and Y are more like Lu and Lr than like La and Ac; and La and Ac are much more like f elements than d elements; and Lr is very weird for a late actinide but seems quite normal for a 6d transition metal. Finally the f block has much better double periodicity as La-Yb: Eu and Yb always show the properties expected for metals which have the half-filled and filled f subshell (viz. Mn and Zn for the d subshell), and Gd and Lu never do. The gas-phase ground-state electron configuration is almost completely irrelevant for chemistry, because not many chemical processes involve gas-phase atoms, and should be ignored.
- Helium. Putting helium over beryllium makes the first-row anomaly trend (the first subshell of any given angular momentum is weird) much better; helium stands in relation to the alkaline earths similarly to how hydrogen does in relation to the alkali metals, looking at atomic properties.
- Strange group numbers. These are inspired by Gary Wulfsberg's Principles of Descriptive Inorganic Chemistry, where he numbers the f groups as 3f through 16f. To avoid confusion with 4f and 5f orbitals I use Roman numerals. Then I extended it to the rest of the elements. The Roman numeral counts the number of valence electrons, the letter tells you which block you are in. The blocks are then classified based on the valence subshells. For p, d, and f blocks you just check the valence orbital with highest angular momentum, which is the most natural thing. For the s block, that was the symmetry break from the idealised left-step table for the very beginning, and besides group 12 are far more like d elements than group 2.
Subject to the constraints of rectangular blocks, these must be the correct form by simple regularity and consistency reasons. Anything else obscures the true electronic basis of the periodic table and is based on local issues (and sometimes non-issues like gas-phase electron configurations) instead of global principles and trends. Ptolemy vs Copernicus, as Droog Andrey (who convinced me back to Sc-Y-Lu) said.
1s 2s 2p 3s 3p 4s 3d 4p 5s 4d 5p 6s 4f 5d 6p 7s 5f 6d 7p
If you want to get any more out of your periodic table than this, I suggest modified Bayley-Zmaczyński (haven't drawn it yet) to elegantly show the following "secondary relationships" pointed out by Droog Andrey.

Sc-Y-La is not taboo, it is a real secondary relationship, just like B-Al-Sc. That's all it should be, I claim. One should not try to promote it beyond what it really is, as that way lies inconsistency.
Conclusion
The physicists have been explaining the periodic system for ages: deriving Madelung's rule (Klechkovsky already did it). Since Hamilton's paper of 1965, they have been explicitly telling chemists about the mistake in group 3 (Lu should be there, definitely not La). Now there is open conflict between physicists and chemists regarding the verification of the superheavies.
It seems to me that physics has through its explanatory power earned its right to assert stronger custody of the periodic table. It is not only the domain of chemists.
(P.S. IRL I am neither a physicist nor a chemist. Even if I do side with the physicists. XD)
As Scerri is now in favour of the left-step table with Sc-Y-Lu and He-Be (see The Periodic Table: Its Story and Its Significance, 2nd ed., p. 403), and he is the chair of the IUPAC project on group 3, I am cautiously hopeful that the story of what I consider the erroneous placement of three elements on the table (helium, scandium, and yttrium) will, soon enough, God willing, have a happy ending.
Acknowledgements
The drawing above and its reasoning have been heavily influenced by copious discussions on WT:ELEM.
I thank most of all Droog Andrey (= A. V. Kulsha) for his correcting me away from the erroneous Sc-Y-La table. I discuss these things mostly with him now.
Special thanks also go to DePiep, Officer781, R8R, Sandbh, and Дрейгорич (Dreigorich) for their discussions since this page was created in 2019, although it should be noted that not all of them agree with everything I say here, most especially Sandbh who still favours La.
Appendix
A list of sources that agree with me that lutetium should take the position under yttrium in the periodic table and provide arguments.
Note, however, that just because an argument is in the sources does not mean it convinces me. The fundamental reason why I am completely convinced that this placement is the correct one is that lanthanum and actinium can use their f orbitals as valence orbitals, but lutetium and lawrencium cannot. This is not quite the argument of any of the sources, although the ones by Hamilton (1965) and Jensen (1982, 2008, 2015) about nonhydrogenic low-lying f orbitals in La-Ac but not Lu-Lr come close. Some other arguments I view as only confirmatory (e.g. property comparisons); some others I am not really convinced by at all (e.g. Scerri 2012; although I agree that the d block should be split, to my mind this requires justification).
- Bury (1921)
- Shemyakin (1932) Zh. Obshch. Khim. 2, 62 (1932) (Russian)
- Landau and Lifshitz (1958)
- Seel (1961)
- Hamilton and Jensen (1963), doi:10.1103/PhysRevLett.11.205
- Hamilton (1965), doi:10.1119/1.1972042
- Luder (1967), doi:10.1021/ed044p206
- Merz and Ulmer (1967), doi:10.1016/0375-9601(67)90527-0
- Chistyakov (1968) Zh. Obshch. Khim. 38, 209 (1968) (Russian)
- Seel (1969)
- Matthias (1969)
- Chistyakov (1970)
- Luder (1970)
- Wittig (1973), doi:10.1007/BFb0108579
- Jensen (1982)
- Holden (1985)
- Fluck and Rumpf (1986), doi:10.1002/ciuz.19860200403 (German)
- Seel (1987)
- Scerri (1991)
- Heyes (1997–8)
- Kulsha (1999?): available on the author's website, pages 1, 2, 3, 4 (Russian)
- Fang et al. (2000), doi:10.1063/1.1322635
- Wulfsberg (2000), Inorganic Chemistry
- Horovitz and Sârbu (2005), doi:10.1021/ed082p473
- Wulfsberg (2006), doi:10.1002/0470862106.ia182
- Ouyang et al. (2008), doi:10.1063/1.2831506
- Jensen (2008)
- Thyssen and Binnemans (2010)
- Scerri (2009)
- Emsley (2011)
- Imyanitov (2011)
- Scerri (2012)
- Bhandari (2013)
- Nelson (2013)
- Scerri (2013)
- Settouti and Aourag (2014), doi:10.1007/s11837-014-1247-x
- Imyanitov (2014)
- Jensen (2015)
- Scerri (2015)
- Settouti (2015)
- Scerri and Parsons (2018)
- Tsimmerman (2018)
- Labarca and González (2019) (Spanish)
- Scerri (2019)
- Scerri (2019)
- Scerri (2019)
- Scerri (2019)
- Tsimmerman and Boyce (2019)
- Alvárez (2020)
- Maeno et al. (2020)
- Scerri (2020)
- Scerri (2020)
{kind=link}
{kind=link}
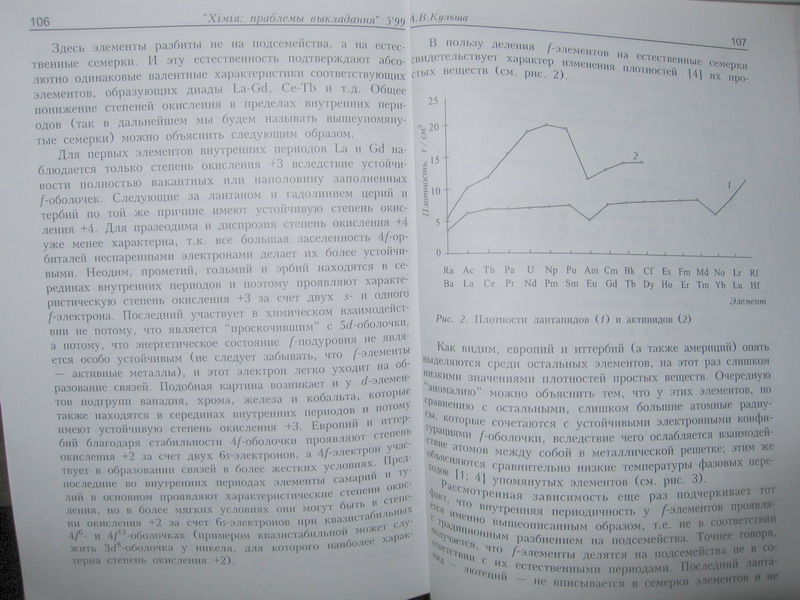{kind=link}
{kind=link}
- Opposition
In support of La there are:
- Smith (1927)
- Trifonov (1970)
- Shchukarev (1974)
- Atkins (2006)
- Lavelle (2008)
- Lavelle (2008)
- Lavelle (2009)
- Restrepo (2017), doi:10.1021/bk-2017-1263.ch005
In support of * there are:
- Xu and Pyykkö (2016)
- Chandrasekar, Joshi, and Ghanty (2019)
Ambiguous:
- Cao et al. (2019). Calls La/Ac and Lu/Lr both d elements.
- Rayner-Canham (2020). Contains whole chapter about this very issue. Writes "Using the long (32-column) form of the Periodic Table, there is no issue: lutetium and lawrencium fall naturally in place". The Luder/Scerri argument is mentioned (Sc/Y/La/Ac breaks the d block); Jensen's arguments appear without criticism, Lavelle's electron-configuration appears but it is also mentioned that ECs are more consistent with Sc/Y/Lu/Lr. However, Rayner-Canham then writes "In this book, ... the lower placements of Group 3 will be left empty" (in order to emphasise continuity over La-Lu and Ac-Lr). Nevertheless on his front page, Lu and Lr go below Y (albeit with a gap so that La-Yb and Ac-No can also be squeezed in).
Not directly related but still gold:
10.1002/anie.197300121
Cites
(fill this up properly)
- Alvarez
- Bent
- Cao et al. (even if they make mistakes about chemical activity of 4f in La, their idea is right)
- Fajans
- Fricke et al.
- Grandinetti (Noble Gas Chemistry)
- Greenwood and Earnshaw
- Grochala
- Gschneidner
- Ionova
- Jensen
- Johnson
- Jørgensen
- Kulsha
- Kulsha again (http://www.primefan.ru/stuff/personal/ptable.pdf, helpful for H and He and many other things here)
- Kulsha and Kolevich
- Nefedov et al
- Ostrovsky
- Pyykkö
- Schwarz
- Seaborg
- Wittig
- Wong
- Wulfsberg
(many are really "et al")
More from Droog Andrey (if only I could understand Russian):